Snakemake Tutorial
Written and presented by Julie Blommaert
What the heck is a snakemake?
Is it a snake that likes to make cake?

It’s a workflow manager:

What is a workflow manager?
Examples of workflow graphs produced by Snakemake:
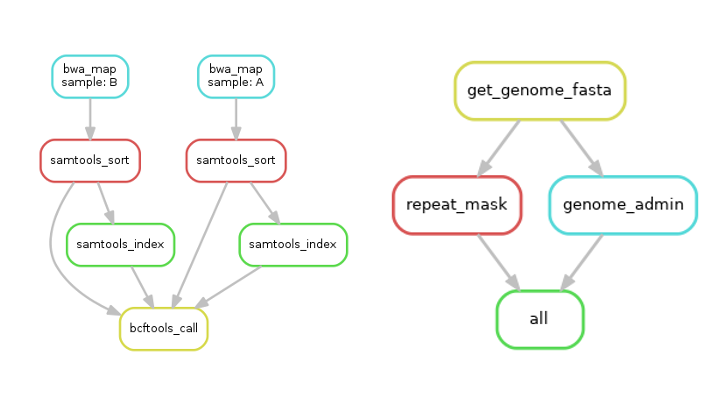
But what’s in it for me?
- Reproducibility
- Not just good for science, but good for future you
- Record keeping
- Easy to share
- Adaptability
How does it work then?
- Each step is called a “rule”
- Each rule is defined by input and output files, and a task
- Snakemake can then figure out how the tasks are connected and if any files are missing or have been updated
- Snakemake builds on Python syntax, but you can use other languages within it (e.g. bash, R)
An example with explanations:
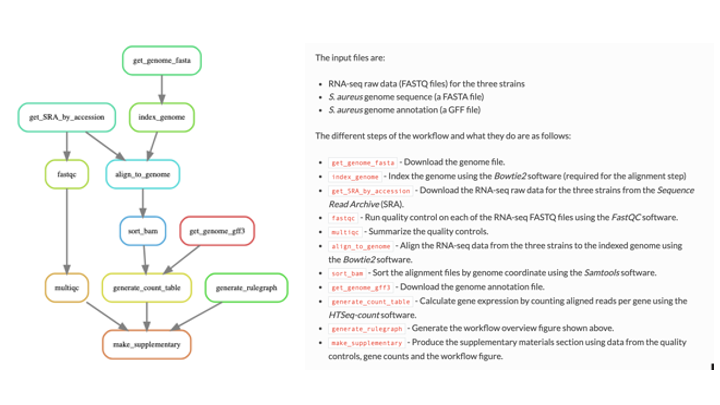
Code example of two ‘rules’:
rule genome_admin:
"""
Shorten the fasta headers,make a blastdb, and fasta index the genome
"""
params:
genome = config["genome_name"]
input:
assembly=expand("assemblies/{name}.fasta",name=config["genome_name"])
output:
index =expand("assemblies/{name}.fasta.fai",name=config["genome_name"])
shell:
"""
samtools faidx {input.assembly}
makeblastdb -in {input.assembly} -parse_seqids -dbtype nucl
"""
rule get_genome_fasta:
"""
Retrieve the sequence in fasta format for a genome.
"""
threads: 1
params:
genomelink = config["ncbi_link"]
output:
outfile=expand("assemblies/{name}.fasta",name=config["genome_name"])
shell:
"""
wget {params.genomelink} -O temp.gz
gunzip temp.gz
cat temp| awk '{{if($1~">"){{printf($1"\\n")}}else{{print $0}}}}'> {output.outfile}
"""
The specific files on which the Snakemake workflow operates are determined by a config file:
config.yml:
genome_name: cygAtr1
ncbi_link: https://ftp.ncbi.nlm.nih.gov/genomes/all/GCF/013/377/495/GCF_013377495.1_Cygnus_atratus_primary_v1.0/GCF_013377495.1_Cygnus_atratus_primary_v1.0_genomic.fna.gz
masking_lib: /proj/sllstore2017073/private/RepeatLibs/lycPyr2_rm2.1_merged.lib
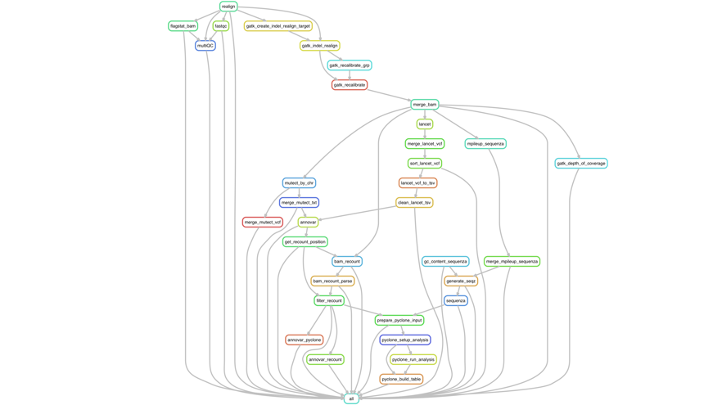
The goal?
a larger workflow:
Links
More comprehensive tutorials:
Many options to organise your workflow
- Bash scripts (you have to start some place)
- Galaxy (web-based workflow)
- Nextflow
- Common Workflow Language (CWL)
