Building blocks
Most bioinformatic methods can be organised into a few basic concepts. I have organised these in terms of how the method influences the sequence files and how it modifies or what information is gathered from them. I use the term ‘sequence file’ very broadly; it can encompass anything from raw reads to assembled contigs to scaffolds to whole organelle or chromosome assemblies. I have furthur divided these into two main classes: lower-level processes and higher-level processes. These are explained in detail below.
Lower-level building blocks
I identify three major types of lower-level processes:
- Pattern searching – finding patterns within a sequence
- Mapping – pairwise alignment of a sequence against a database
- Clustering – group together a collection of sequences based on identity
Each of these process types can encompass or be a part of many programs, and be applied to multiple sequence types–as outlined above. Pattern searching is typical of early steps in most processing of raw reads, and includes adapter and primer removal, as well as quality filtering. However, it also is involved in many downstream processing steps, including searching for coding regions or CG islands on assembled contigs, or repeats across whole genome assemblies. Note that often these searches are looking for patterns based on existing references, such as adapter sequence files or repeat databases, but they can also involve looking for patterns not neccesarily dependent on any external reference, such as start and stop codons along a contig.
For Mapping processes, these involve searching whole sequences against some kind of reference database. As with Pattern searching, these can involve a BLAST search of short reads or assembled contigs or aligning RNA reads to a transcriptome. Kraken, HMM
These first two processes may seem like the same thing, and indeed they are more or less two sides of the same coin. But because they are usually very separate in pipelines I am keeping them distinct. I distinguish them by what is the subject and what is the object of the search: generally Pattern searching is looking for elements of a database somewhere along a sequence (database is subject, sequence is object), wherease Mapping is searching in a database for whole sequence reads (sequence is subject, database is object). Though for many searches, especially local alignments, matches to only part of the sequence may be found, it is usually the whole sequence that is the search query. Because of the nature of both of these (especially where pattern searching is looking for shorter and often, fragmentary patterns), generally the algorithms used for both are different.
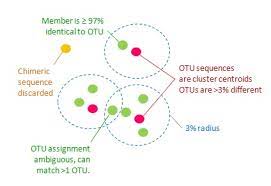
The third type, Clustering, involves grouping or clustering a collection of sequences based on similarity. It is only comparing within a group of sequences, and does not generally involve any external references (of course, there are many hybrid clustering approaches that combine mapping and clustering, but often these happen in turn). Clustering methods are found across many bioinformatic pipelines and include kmer counting, OTU clustering (in metabarcoding), and dereplication. I include genome assembly here, though genome assemblies usually involve a wide range of steps, but building larger contigs from alignments of kmers is one of the core approaches. I also include multiple sequence alignment (MSA) here; while it is an alignment method, it is distinct from the Mapping alignment methods described above, as those generally involve pairwise comparisons–aligning a single query separately to each of many subjects in a database. While a MSA is generated from the simultaneous alignment of multiple sequences–at heart it is a very different approach.
Here is a summary table that includes some examples of each method:
| Pattern searching | Mapping | Clustering |
|---|---|---|
| finding patterns within a sequence | pairwise alignment of a sequence against a database | group together a collection of sequences based on identity |
| adapter/primer removal | align reads to genome | kmer counting |
| gene prediction | BLAST search | genome assembly |
| motif searching | HMM search for protein domains | dereplication |
| repeat finder | create OTU table | multiple sequence alignment |
 |
 |
 |
Next level: higher order processes
From the basic processes, there are higher level methods that make statistical inferences about the results of the lower-level building blocks. For example, a phylogenetic analysis will try to infer relationships among samples in a multiple sequence alignment. Likewise, a principal component analysis (PCA) is often used to determine relationships among Operational Taxonomic Units (OTUs) resulting from a clustering analysis, such as USEARCH. I also include variant calling algorithms, such as GATK Haplotype Caller and FreeBayes, in this category, as they make inferences about statistically robust Single Nucleotide Polymorphisms (SNPs) and other variants resulting from mapping reads to a genome or transcriptome.